Copy Number Variants with Shallow Sequencing
- January 1, 2016
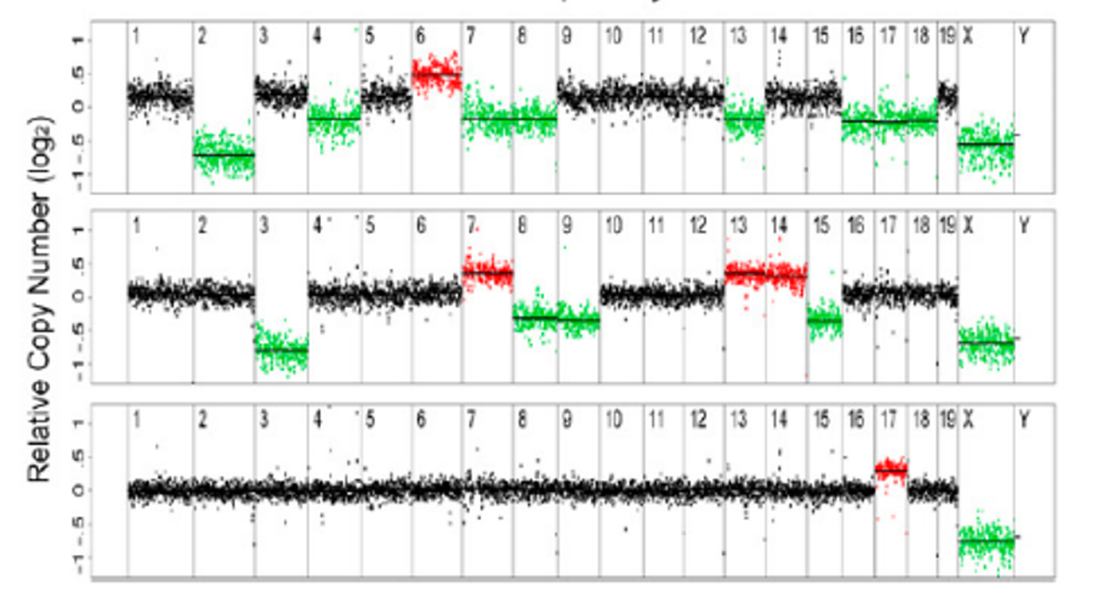
While working at MIT, Kristin Knouse (Amon Lab) and I developed a workflow to reliably detect copy number variants (CNVs) from shallow whole-genome sequencing data. Our goal was to investigate previous reports of unexpectedly high levels of large-scale CNVs in somatic neural cells. Through simulations, we defined optimal parameters for CNV detection and found that, using these optimized parameters, the prevalence of large CNVs in somatic cells is lower than previously reported, which aligns better with biological expectations. This work established a standard for calling CNVs from shallow whole-genome sequencing data in single cells.
Subsequently at Philips, in collaboration with Timour Baslan at MSKCC, I refined the algorithm to analyze shallow whole-genome sequencing data from tumors by incorporating adjustments for sample purity and ploidy. Our aim was to evaluate whether this approach could serve as an alternative to traditional methods such as cytogenetics and SNP arrays, and to assess its benefits over targeted sequencing in clinical settings. We benchmarked the method using both hematological and solid tumor samples. With optimized parameters and workflows, we concluded that megabase-scale CNVs can be reliably detected using shallow WGS at coverage as low as 0.1x. However, due to its limited depth, shallow WGS cannot detect loss of heterozygosity (LOH), inversions, or translocations. As sequencing costs continue to decline, WGS is becoming increasingly accessible and holds promise to replace conventional cytogenetic techniques in clinical practice.
- Kristin A Knouse, Jie Wu and Angelika Amon. Assessment of megabase-scale somatic copy number variation using single cell sequencing, Genome Research, 2016. gr.198937.115. Link
- Kristin A Knouse, Jie Wu, Charles A Whittaker and Angelika Amon. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proceedings of the National Academy of Sciences, 2014. 111(37):13409-13414. Link
- Kristin A Knouse, Jie Wu, Austin Hendricks, Detection of Copy Number Alterations Using Single Cell Sequencing, JoVE, 2017. 120: e55143–e55143

